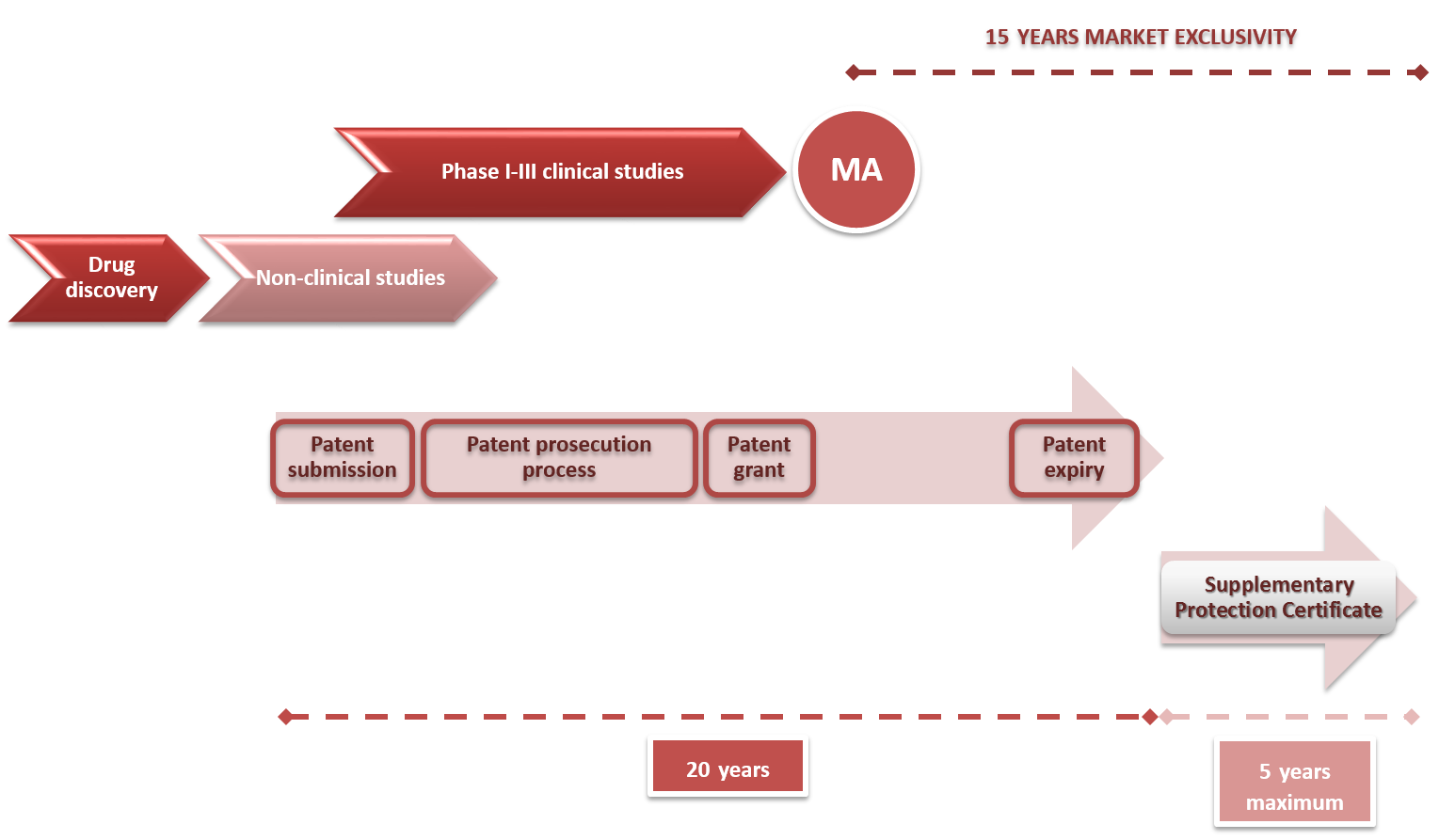
La solicitud de una patente otorga al solicitante el derecho de exclusividad de mercado durante 20 años. Esto no siempre es exactamente así, pues cuando se solicita una PCT (patente internacional) que proviene de una solicitud prioritaria nacional, la protección efectiva será de 21 años (20 años desde la solicitud de la PCT), y si nos referimos a una patente biotecnológica o farmacéutica, el asunto se complica mucho más.
En el sector farma-biotech, dos legislaciones aparentemente tan dispares como las leyes de patentes y el marco regulatorio, van totalmente de la mano. En otros mercados, el derecho de exclusividad puede comenzar en el mismo momento en que se presenta la solicitud de patente, pero en el caso de una patente farma-biotech, su comercialización está supeditada al cumplimiento de una serie de requisitos regulatorios, y no es hasta la obtención de la autorización de comercialización cuando se puede disfrutar de este derecho de exclusividad.
Normalmente, la patente se solicita en las primeras fases de desarrollo, tras la etapa de drug discovery y con unos primeros datos en animales, y debido a los tiempos necesarios para finalizar los estudios clínicos y no-clínicos, que en muchos casos se dilatan debido a problemas de financiación, el periodo de 20 años de vigencia de una patente, puede convertirse en sólo unos pocos años de protección efectiva, que no sería suficiente para asegurar el retorno de la inversión hecha en las etapas de investigación.
Por ello, tanto en el sistema americano de patentes como en el sistema europeo, existen certificados complementarios de protección (conocidos como Patent Term Extensions y Supplementary Protection Certificates respectivamente, y como Certificados Complementarios de Protección en España) que proporcionan hasta 5 años adicionales de protección, para garantizar un mínimo de 15 años de exclusividad de mercado a partir de la autorización de comercialización.
En el caso de Europa, estos certificados deben solicitarse en cada territorio individualmente, dentro del plazo de 6 meses tras la primera autorización de comercialización en la Comunidad Europea; sólo puede solicitarse un certificado por cada patente base, aunque la patente proteja más de un producto, y se limitan a un CCP por producto, por lo que una patente de segundo uso médico no podría solicitarlo si el producto ya ha recibido previamente una autorización de comercialización.
Además, este certificado podría prorrogarse por 6 meses adicionales, en el caso de medicamentos pediátricos. Por todo ello, la gestión de la solicitud de los CCPs es crucial para la estrategia de la compañía y deben estudiarse todas las opciones cuidadosamente.
Cabe destacar también una figura existente en el mercado americano, que hasta ahora se ha quedado en algunas propuestas en Europa, denominada Priority Review Voucher, incluida entre los incentivos para el desarrollo y comercialización de medicamentos huérfanos. Este voucher se traduce en un trato prioritario en la tramitación de la autorización de comercialización, adelantando así el lanzamiento al mercado del producto y por tanto puede suponer un mayor tiempo de exclusividad de mercado, siempre sin sobrepasar los 20 años de vigencia de la patente o los 15 años efectivos en el caso de solicitar un SPC.
Lo más curioso de los PRVs es la posibilidad de utilizar esta prioridad ante las autoridades regulatorias para otro medicamento que no sea el designado como huérfano, e incluso, la posibilidad de vender el PRV a otra compañía. En esta línea, se ha propuesto una figura bastante más polémica, las Wild-Card Patents, que proponen directamente una extensión en la duración de una patente para aquellas empresas que lleven a mercado un medicamento huérfano, e igualmente sería un derecho transferible, lo que supondría un cambio importante en las reglas del juego.
Como suele ocurrir en materia de patentes, la respuesta a nuestras preguntas no suele ser sólo una, y su tiempo de vigencia no es una excepción.
Se me hizo difícil comprender el argumento «No podemos recuperar beneficios porque hemos invertido mucho en investigación» cuando descubrí que, en general, invierten más en marketing que en i+d.
Además, entiendo que hay muchas tácticas para adquirir altas cuantías de ingresos a raíz de crear nuevas patentes con poca inversión/esfuerzo. Un ejemplo son los Me Too. Eso sin mencionar el hecho de lo POCO que le cuesta a la industria adquirir un principio activo que ya haya sido sesgado como interesante y estudiado en sus fases primarias a costa del dinero público de, por ejemplo, universidades y otras muchas estrategias que ni conoceremos…
Hay amplia bibliografía en internet, pero acá un resumen para leer en un momento:
http://farmacriticxs.ifmsa-spain.org/esp/descargas/MedicamentosMeToo.pdf
Hola Isa, muchas gracias por tu comentario.
El tema de los me too, también conocido como evergreening, es sin duda una práctica tristemente extendida en la industria farmacéutica, pero no creo que el método de control más efectivo para evitar estos abusos sea modificar las leyes de patentes y mucho menos deslegitimar el derecho de todo inventor a disfrutar de su periodo de exclusividad a cambio de la divulgación de su investigación. No olvidemos, que las patentes no son usadas sólo por multinacionales, y que son el único arma con el cuentan los centros públicos de investigación o empresas biotech para hacer rentable su inversión en investigación.
Una patente sólo llega a ser concedida cuando cumple los requisitos de novedad y actividad inventiva; la mejora terapéutica, por el momento, no está incluida como requisito de patentabilidad, ¿quizás debería estarlo en el caso de los fármacos?, pregunta compleja desde luego.
En esta línea, tenemos el reciente caso de Novartis vs Ley de Patentes India, que el tribunal supremo ha fallado a favor de los segundos en base al artículo 3d de la Ley de Patentes India, por considerar que la patente que protege Glivec no es suficientemente innovadora para gozar de esta exclusividad de mercado.
Sin entrar a debatir en si la sentencia es correcta o no, principalmente porque no conozco el caso tan a fondo para entrar en este debate, sólo una pequeña reflexión: “Si realmente el producto de Novartis no aporta ventaja alguna respecto a los productos que ya se encontraban en el mercado, ¿qué interés puede tener la industria genérica india en producirlo, infringiendo una patente que aún se encuentra vigente, en lugar de producir otros fármacos equivalentes cuya patente ya ha espirado?”
Como ya he dicho, la solución no está en un endurecimiento de los requisitos de patentabilidad, de hecho, el mecanismo de control que considero clave para evitar que “falsos innovadores” lleguen a disfrutar de un nuevo periodo de exclusividad, se encuentra citado en el artículo que nos adjuntas: prescripción informada en base a estudios coste-beneficio.
Personalmente, creo que un estudio farmacoeconómico debería adjuntarse obligatoriamente a toda solicitud de registro, algo que ya ocurre en otros países como Australia, Canadá o Reino Unido, y que quiero pensar se irá extendiendo a otros territorios. Pero en cualquier caso, una vez el fármaco se encuentra en el mercado, será un prescriptor bien informado el que pueda decidir de manera crítica cuándo recetar el fármaco «innovador» y cuándo el genérico del fármaco del que procede ese me too.
En este punto entiendo que interviene el papel «informativo» de la inversión en marketing de la empresa farmacéutica que citas al principio de tu comentario, pero me temo que contra eso, sólo nos queda confiar en el conocimiento y ética del prescriptor.