Durante los últimos meses, no han sido pocos los eventos, presenciales y/o virtuales, en torno a la aplicación de la nueva regulación sobre ensayos clínicos, tras la entrada en vigor del Real Decreto 1090/2015.
El pasado viernes, Bioclever me invitó a participar en una mesa redonda cuyo enfoque me pareció algo diferente al de los otros encuentros, y mi papel era el de moderar el debate final y el turno de preguntas del público, tras exponer las conclusiones de la jornada. Por supuesto, no pude decir que no al reto de sacar conclusiones de ponencias cuyo contenido desconocería hasta ese momento… 🙂
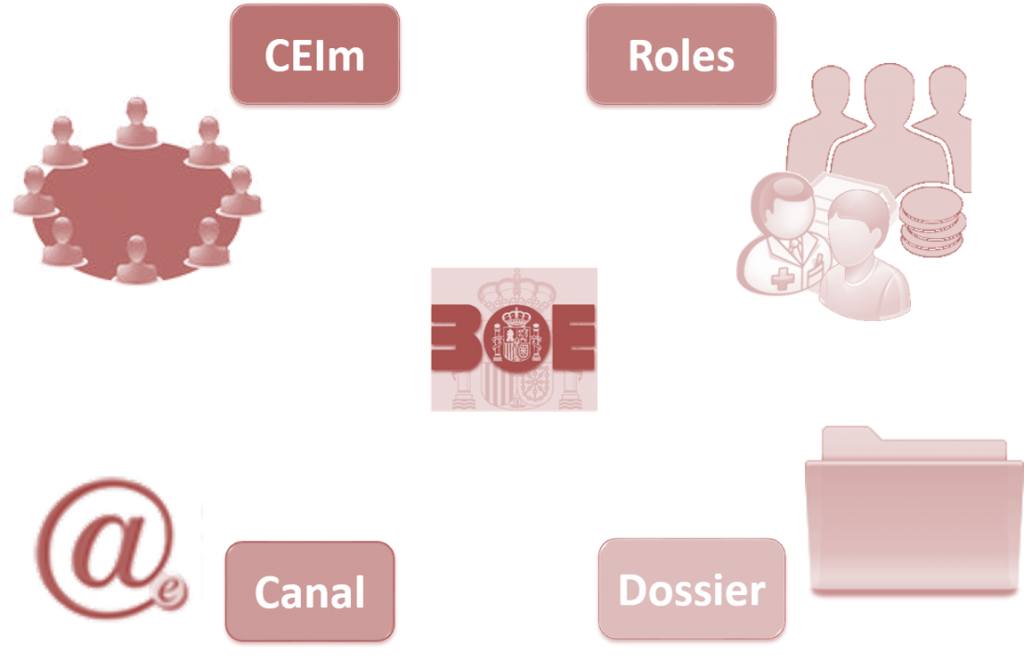
Como soporte a mi intervención llevé la imagen que he usado de cabecera de este post, y por suerte, mis dotes de vidente funcionaron y los ponentes que me precedieron orientaron sus ideas sobre el nuevo Real Decreto en la misma línea que las mías.
Desde mi punto de vista, el cambio más obvio y el que ha generado más expectación en lo referente a su implantación en la práctica, ha sido el nuevo concepto de CEIm (comité ético de investigación con medicamentos) que vendrá a sustituir a los antiguos CEIC (comité ético de investigación clínica). La evaluación de la solicitud de ensayo clínico por un único CEIm (que será elegido por el promotor, entre la lista de CEIm acreditados por la AEMPS), con el consecuente pago de una única tasa, y la emisión de un único dictamen para todo el territorio, se ha presentado como la gran apuesta del Real Decreto para reducir la burocracia y acelerar los trámites.
El contenido del Dossier para la solicitud de ensayos clínicos se mantiene más o menos igual en el fondo, aunque sí tiene novedades en la forma, principalmente para permitir la evaluación simultánea por parte del CEIm y la AEMPS, y con la voluntad de que en un futuro será presentado una única vez para toda Europa, sin duda un avance considerable para los ensayos clínicos multicéntricos internacionales. Y no nos olvidemos del futuro modelo de contrato único con los centros, que mantenemos la esperanza de que sea posible, aunque seguro que sea cuál sea su formato, no contentará a todos…
Y en línea con esta agilización de los trámites, la promesa de un canal de comunicación vía electrónica, y bidireccional, que se incorpora, al menos en parte, en nuestra legislación nacional. La presentación y modificación de toda la documentación a través de la sede electrónica de la AEMPS (parece que el portal único europeo que reza en el Reglamento aún tendrá que esperar…), el registro obligatorio de todos los ensayos clínicos (no olvidemos que pueden ser con medicamentos o con productos sanitarios en investigación) en el Registro Español de Ensayos Clínicos, la notificación de efectos adversos a través de Eudravigilance_CTM mediante una única comunicación a nivel europeo (eliminando las notificaciones a los CEICs), y con el compromiso de crear un portal donde se harán públicos los efectos adversos de medicamentos…
Pero mi lectura particular sobre los objetivos y el trasfondo de este nuevo Real Decreto es la adaptación de la legislación europea, y por ende de la española, a un cambio de roles en la investigación clínica. En mi opinión, el escenario en que el promotor controlaba y pagaba, el investigador acataba y el paciente firmaba, ha pasado a la historia, y en este nuevo ecosistema de innovación cada uno de los actores está aún buscando su papel.
Ya no sólo las big farma esponsorizan ensayos clínicos, podemos encontrarnos con promotores de lo más variopinto: pequeñas y medianas empresas de diferentes ámbitos (como spin-off universitarias u hospitalarias), fundaciones, investigadores independientes, grupos corporativos o incluso asociaciones de pacientes, que financian y ponen en marcha un protocolo clínico diseñado en base a las necesidades que han detectado en su particular realidad.
El paciente empoderado tiene cada vez un papel más proactivo en la práctica clínica, y era sólo cuestión de tiempo que reivindicara su lugar también en la investigación, estando ahora, por ley (o por decreto), directamente implicado en la evaluación de los ensayos clínicos, con la incorporación de al menos un representante de los pacientes en los CEIm.
Sin duda, la medicina basada en la evidencia, que ha guiado (y debe seguir guiando) la investigación clínica, está más cerca que nunca de la medicina centrada en el paciente, y ni la práctica clínica ni la investigación pueden ya interpretarse si no es teniendo en cuenta ambos enfoques: poblacional e individual.
Pero cuando nos movemos en terrenos movedizos, no hay leyes que puedan regular lo que la realidad nos demanda, y el papel de los hospitales como motor de innovación y pieza clave para la incorporación del conocimiento generado en el Sistema Nacional de Salud, sigue a mi parecer sin contar con los medios que debería.
La declaración de intenciones que este Real Decreto hace en lo referente a los ensayos clínicos no comerciales (aquellos propuestos por investigadores independientes), se queda coja al no tener reflejo en unos presupuestos concretos para el impulso de dichas investigaciones, así como de plataformas de apoyo para su gestión. Y ese nuevo concepto de «ensayo clínico de baja intervención» que deja entrever una legislación más laxa en pro de facilitar su desarrollo, pero sin dejar claro cómo garantizar la calidad y los derechos de los pacientes involucrados, me deja algo dudosa…
Sin más, como comentaba con un colega al terminar la jornada, seguiremos con expectación cómo se van desencadenando los acontecimientos, cómo se van implementando las novedades de este Real Decreto y cómo se resuelven las incógnitas que deja sobre la mesa…